жЛашвЛВНЃЌПьЫйПЊЪМ
ЮЂаХЩЈвЛЩЈЃЌПьНнЕЧТМЃЁ
ЗДГхСІ:
2025-1-25 21:01 [ЛиИД][0]
yuling_pg: МсГжСЫЪЎЮхФъРВ
2024-12-19 15:25 [ЛиИД][0]
ЬњТэН№Иъ: бЇЯАЃЁМггЭ
2025-1-9 11:08 [ЛиИД][0]
УЈпфЕФКњзг: НјВНвЛЕуЕу
2024-9-1 18:36 [ЛиИД][0]
ЧѓжњЃКЮвдѕУДжЄУї Юв
2дТ18Ше 19:00-21:00
ЫежнДѓбЇИНЪєЕкЖўвНдК
2дТ16Ше 14:00-17:00
ЫежнЪаСЂвНдКЬЧФђВЁЁЂ
ЪЙгУЕРОп ОйБЈ
БОАцЛ§ЗжЙцдђ ЗЂБэЛиИД





 ТГЙЋЭјАВБИ 37020202001532КХ
ТГЙЋЭјАВБИ 37020202001532КХ 
 бЇЯАЃЁМггЭ
бЇЯАЃЁМггЭ

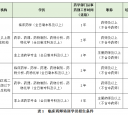




 IPПЈ
IPПЈ ЙЗзаПЈ
ЙЗзаПЈ

 ЬсЩ§ПЈ
ЬсЩ§ПЈ жУЖЅПЈ
жУЖЅПЈ ГСФЌПЈ
ГСФЌПЈ ањЯљПЈ
ањЯљПЈ БфЩЋПЈ
БфЩЋПЈ ЧРЩГЗЂ
ЧРЩГЗЂ ЧЇНяЖЅ
ЧЇНяЖЅ ЯдЩэПЈ
ЯдЩэПЈ