|
最近的“冰桶挑战”越来越火,甚至扩展到了政界和娱乐界。而在国内,人们在享受这场全民狂欢的同时,却好像忽略掉它背后想要传达的信息。我想,这并不是发起人的初衷。 两个月前,我在广州看了一场“罕·见的世界——罕见病家庭纪实摄影展”,来自一批优秀摄影师,历时3个月走进20个罕见病家庭,用照片和文字跟踪记录他们的日常生活,并希望以展览的形式唤起人们的注意。
可惜,我去看的时候,参观者寥寥无几,捐赠者更少。 其实,我们是不是在冰桶狂欢之余,稍微花一点时间真正地去关注和了解这些罕见病呢?
这个摄影展的下一站是西安,9月;再下一站是成都,11月。 展览有一句Slogan说“在死亡面前,我们都是幸存者。” 如果你真的为这份幸运想要做些什么,可以考虑去观展,也可以点击“阅读原文”,去他们的义卖区看看。有5块钱的徽章,25块钱的笔记本,68块钱的帆布袋,88块钱的T恤。 当然,你也可以把这篇文章转出去,让更多的人真正关注罕见病,我们不该只知道冰桶和ALS对吧。
瓷娃娃的人生不易碎 图文/杨宽 
由于不能运动,为了增强自己抵抗力,张金生一年四季坚持用冷水洗头,避免生病为别人带来不必要的麻烦。
1954年出生的张金生出生不久身体就发生骨折,在他的记忆里,小时候身体发生的骨折就像吃家常便饭一样。 张金生清楚地认识到自己是“成骨不全症”已经是2007年的事了,那年他53岁,通过看报纸为自己确诊。成骨不全症患者被称为“瓷娃娃”,因先天遗传性缺陷,导致骨质薄脆,打喷嚏、翻个身都可能骨折。 现在的张金生下身完全萎缩瘫痪,上肢也只有经过多次骨折弯曲的左臂可以活动。 会电焊的弟弟为他焊制了一辆带有转向轮的铁车,每周弟弟都会带他出来在小区外透透风,但一定是在黑天的夜晚,因为在夜幕的掩盖下很少会有他人的异样眼光。 尽管张金生长期闭门在家,但他的思想并不闭塞,身边有支口琴。在电脑的一个音乐软件上还录有他唱的30多首歌,这其中有民族风格的《父亲》,更有流行风格的《菊花台》。他说音乐是有生命的东西,总有力量在里面奔涌。
成骨不全症(OI)
又称脆骨病,发病率1/10000~1/15000,是一种先天性遗传疾病,表现为骨质脆弱、蓝巩膜、耳聋、关节松弛,目前临床上用于治疗该症的双磷酸盐类药物虽在医保目录里,但该药的适应证里并未列明成骨不全症,因此患者需自费。该群体也被称为“瓷娃娃”。
月亮孩子的现实和理想 图文/钟锐钧 
记者问她:“你有想过谈恋爱吗?”她回答得很快:“当然有啊!” 高洪舟今年22岁,同事和朋友都称呼她为小仙女。 用白皙来形容高洪舟可能都不够准确,雪白更贴切些。她患有白化病,全身雪白,头发也是银白色的,眼睛像外国人一样,颜色很浅,仿佛心里想什么,一眼便能看透。 3岁的时候,小小的高洪舟就发现了自己异于常人:很怕光,皮肤也比一般的孩子白得多,妈妈开始给她染发,一直到18岁。 18岁那年,高洪舟决定不再染发了。一是染发对人体有伤害,二是她觉得自己开始认同作为一个白化病人的身份。同一年,高洪舟考上渤海大学公共事业管理专业,她选择专业的目的很明确:帮助残疾人和罕见病病人。 白化症(Albinism)
发病率约为1/15000,是一种常染色体隐性遗传病,表现为眼睛视网膜无色素,虹膜和瞳孔呈现淡粉色,怕光,低视力,眼球震颤,看东西时总是眯着眼睛。皮肤、眉毛、头发及其他体毛都呈白色或白里带黄。目前仅能通过物理方法,尽量减少紫外辐射对眼睛和皮肤的损害。该群体也被称为“月亮孩子”。
“不食人间烟火”的孩子 图文/肖翊 
瀚林爸爸专门组建了一家为PKU孩子提供食物的公司,目前已在全世界收集了两百多种适合PKU患者使用的食品。
王瀚林,是一名PKU孩子,今年5岁半的他在北京市丰台区的一家民办幼儿园上大班。暑假里,瀚林和姑姑家的两个孩子一起在奶奶家。奶奶有规定要避开瀚林的视线躲到厨房里吃冰棍。一天,二姑回家接孩子发现王瀚林蹲在厨房的地上,正在舔扔在垃圾桶里的冰棍包装袋上的汁。瀚林发现二姑,他忙解释:“我就看看。” PKU孩子因肝脏中缺乏一种酶,不能充分分解蛋白质中的苯丙氨酸,而苯丙氨酸在体内蓄积过多,将衍生成一系列毒性物质,伤及大脑细胞,使人智力残疾。蛋白质是绝大多数食物中不可或缺的营养成分,而苯丙氨酸就存在于蛋白质之中。因此,PKU孩子只能食用人造的无(低)苯丙氨酸食品。 “5年了,每天一睁眼就开始为瀚林的三餐发愁”,瀚林妈妈说。因为PKU孩子一日三餐必须吃特制食品,不管是幼儿园还是学校,一听说孩子吃饭特殊,都连声拒绝道:“负不起这个责任”。 苯丙酮尿症(PKU)
在我国发病率为1/16500,是一种先天性遗传代谢病,表现为智能低下,惊厥发作和色素减少。目前低苯丙氨酸饮食疗法是治疗经典型苯丙酮尿症的唯一方法。该群体也被称为“不食人间烟火的孩子”。
遥远又近在咫尺的幸福小区 图文/李昊 
韩硕平时只穿宽松的衣服来掩盖肿大的腹部。
韩硕今年8岁,爸爸韩旗在离家不远的饭店掌厨,妈妈留在家里照顾刚出生的小弟弟。2009年,韩硕的大舅抱着当时3岁的韩硕玩耍时,发现腹部有个硬块,几经若干家大医院诊断,最后被确诊为罕见病戈谢病。 戈谢病为常染色体隐性遗传疾病。韩硕的父母由于均为携带者,因此生小孩患病的可能性为1/4,很不幸,老大韩硕就成为了戈谢病患者。今年,韩旗夫妇的第二个孩子出生,小家伙是个男孩。在老二出生前,韩硕的妈妈在协和医院接受孕前检查,得知孩子不是戈谢病患者才继续生产。 现在,韩旗最怕韩硕突然流鼻血。9月开学前,韩硕连续4天流鼻血,其中一天狂流4小时,家里的大盆接到满。 戈谢病
在我国发病率为1/200000~1/500000,是一种溶酶体贮积症,表现为多系统的脂质沉积,累及骨髓、肝脾、骨骼及神经系统,会导致疼痛、骨损伤甚至死亡。目前治疗I型戈谢病最有效的方法是葡糖脑苷脂酶(伊米苷酶注射液)替代疗法,此药已在我国药监局注册并上市,但费用昂贵。
困难是男子汉的勋章 图文/吴皓

李河言兴趣爱好很广,曾学过跆拳道、书法、绘画和声乐。由于身体原因,已经放弃了跆拳道,但仍坚持学习绘画和声乐。
李河言(小名龙龙)2010年开始被诊断出患有黏多糖贮积症,母亲带着他去过上海北京等医院就诊都效果不明显,目前只能通过每天接受按摩治疗来缓解肌肉萎缩等症状。 龙龙每天的按摩分为两部分,针灸(20分钟)和全身揉搓按摩(大概半小时);针灸时需要在头部和背部很多地方扎针,他说刚扎的时候会有点点疼,不过久了也会习惯,扎完针只能面朝下躺着,有时候会很无聊,只能用看手机玩游戏打发时间,不过诊所护士为了防止他弄到针也经常会“没收”手机,之后龙龙就只能很郁闷地躺着,四顾张望。 由于龙龙妈妈忙,龙龙很多时候是自己一个人去诊所做治疗。龙龙的心态很好,即使忍受着疾病带来的困扰,他也还是会和身边的人开玩笑,诊所的医生们也把他当成一个朋友而不是病人来看待。 龙龙到过国内很多地方旅行,包括香港台湾,海南岛,上海北京等,他自己说他最喜欢上海,他很喜欢到各地去尝不同风味的小吃。
黏多糖贮积症(MPS)
在北美和欧洲的总发病率约为1/25000,是一种溶酶体贮积症,表现为粗糙面容、角膜混浊、关节僵硬、身材矮小、肝脾增大、耳鼻喉部病变等。目前国外已经有针对黏多糖贮积症某些亚型的酶替代治疗药物,但这些药物还未在国内上市,即使上市,每年高达百万元的费用且需要终身用药,若无医疗保险支持大部分家庭也无力承担。该群体也被称为“黏宝宝”。
呼吸,仅为活着 图文/吴宏 
李文峰在吃午餐,桌上摆着他最感兴趣的投资理财书,他希望能学习投资理财知识,帮家里分担一些经济压力。
呼。。。呼。。。有节奏的声音在一间十几平米的小屋里仿佛被放大了一般听起来格外清晰,声音来自一台呼吸机,它源源不断地通过几米长的空气导管顺着李文峰气管切管处把空气压送到他的胸腔内。 对于这位二十四岁的小伙子来说,这是他生存下去的唯一依靠。作为这个家庭唯一的男孩,李文峰2011年被确诊为患有庞贝病,更加不幸的是,他的大姐,1982年出生的李文文和1984年出生的二姐李静也同时被确诊患有庞贝病。
时至今日,李文峰依旧要靠呼吸机24小时不间断的维持呼吸,农村常见的时而停电成为了李文峰生命的最大威胁。 天气好的时候,李文峰的母亲宋玉兰会费力的把儿子从床上搬到轮椅上,附带着几十米的电源线连接好维持李文峰生命的呼吸机和吸痰机,为他泡上一壶热茶,让他一个人静静的坐在院子里,奢侈的感受一下蓝天白云。
庞贝病(Pompe disease) 发病率约为1/40000,是一种溶酶体贮积症,婴儿型患者表现为无力、心脏扩大等,晚发型表现为进行性肌无力。美国开发出来的针对该疾病的特效药物仍未在中国上市。国内已有的庞贝病患者,通过香港等途径外购药物维持治疗,每年的花费需在200万元左右。 活着,在无奈和坚持的边缘 图文/梁莹菲

晚饭后,嘉璐要打一个饱嗝,不然打进胃里的东西容易被吐出来,璐妈把嘉璐的身子支起来,等待着这个饱嗝。
嘉璐有点像童话书中描述的小精灵,身体过分瘦小让她的头显得很大,脸色异常苍白,嘴唇倒是好看的粉红色,而最吸引我的是那双大眼睛。从我走进小屋的那一刻开始,那双眼睛便紧盯着我这个外来入侵者不放。 我站在门口呆呆地和这个女孩儿对视良久,正当我向前几步准备靠近她时,她的眼睛突然瞪得很大,转头盯住天花板,手脚不约而同地紧缩起来,嘴巴发出了痛苦而含糊的叫喊声,璐妈这时立刻冲过来抱住她,把她的头摆正,嘴里念叨着“不抽了,宝贝不抽了……”过了一段时间,嘉璐终于停止了抽搐,而刚刚在她眼睛中的灵魂仿佛被抽空,眼珠子呆呆地垂在一侧,身体也软了下来。 “没吓着你吧?她刚刚癫痫发作了。”璐妈微笑着问我。
尼曼匹克最典型的症状是吞咽困难,嘉璐不仅咽不下去一点饭,就连自己的口水也很难咽下去,在唾液或痰积累到一定程度时,就会被呛到咳嗽不止,咳到嘴唇发紫,眼泪狂飙,此时一定要有人及时帮她用注射器把唾液抽走。 尼曼匹克病(NPD) 发病率约1/25000,是一种常染色体隐性遗传性疾病,表现为肝脾肿大,亦有神经系统损害。目前尚无特效疗法。
破茧而出的蝴蝶宝贝 图文/孙炯 
妈妈在点评周密的素描作业,从2012年开始,周密对绘画产生兴趣,父母认同这项爱好,并希望绘画能发展为周密的谋生手段。
2003年8月,周密出生在一家妇产医院的特需产房。一出产道,助产士呀一声,对边上陪护的父亲周迎春讲:“你看,不是我弄的。”卧产床的母亲徐玲急了,问怎么了。周迎春用平稳的口气安慰:“没什么,小问题。”徐再追问,他只好答:孩子腿上的皮没长好。 出院的第二天,上海瑞金医院确诊她为大疱性表皮松懈症(简称EB)。这是一种罕见的遗传病,不传染,但表现为皮肤和其它有关组织结构脆弱,反复长疱。 彼时,瑞金医院的皮肤科主任私下说养不活,拿来一本书,对照着讲一些护理建议,让父母抱回家了。有一回,在求医路上,徐玲对周迎春说:“只要孩子脑子没事,能写字,能走路,我就满意了”。 现今,除了每日早晚要检查身体,处理伤口,替换敷料、纱布外,母亲徐玲觉得,相比普通孩子,周密已经没什么不同了。 大疱性表皮松解症(EB) 新生儿发病率约为1/50000,是一种遗传性皮肤疾病,表现为皮肤、眼睛、口腔和食道等粘膜组织在受到轻微摩擦或没有明显原因的情况下会发生水疱或血疱,进而导致创伤溃烂。目前尚无特效疗法。该群体也被称为“蝴蝶宝贝”。
生命很慢,无力也是一种有力 图文/王中杰 
早上起床后手没力扎不起鞭子,阿在就把肘部搭在书架上做支撑,一样能扎起漂亮的马尾,如果实在扎不起来就用发夹夹着。
郭云在家的木屋因腾冲的雨季变得潮湿,这天中午她感觉特别累,吃过午饭拄着拐杖慢慢爬上二楼,打算躺在床上休息一下。 换睡衣对她来说有点困难,所以她只是把鞋脱掉,然后坐在床上,用双手分别把自己的左脚和右脚抬到床上,最后慢慢把床尾的被子拉到身上,一头仰到床上,睡了下去。 郭云在是一位重症肌无力患者。 阿在说生病让她错失了很多东西,与外面的世界隔绝久了,阿在很想走出小镇。她最大的梦想是病好起来,实现生活自理。这样就能做很多喜欢的事情。 病前病后,阿在一共当了八年多乡村小学教师。现在,她在看教育学和心理学的书,说想用这些专业知识,再结合上自己之前的经验,给乡村小学教师提供一些教育经验,她希望未来能在这方面努力。 重症肌无力(MG) 发病率约为1/12500,是一种自身免疫性疾病,表现为眼睑下垂、饮水咳呛、吞咽困难、表情僵硬、四肢无力,甚至累及呼吸肌,引发危象,危及生命。目前尚无特效疗法,经休息和胆碱酯酶抑制剂治疗后可减轻症状。
生活让我如此歌唱 图文/吴宏 
刘杨每周去几次基督教堂,在唱诗班上跟随着和她同龄的正常年轻人一起歌唱。
31岁的刘杨除了去过北京看中医治病外就一直待在位于山东省青岛的家,这个出生于1982年的年轻人在1998年被诊断患有多发性硬化症,一种慢性、炎症性、脱髓鞘的中枢神经系统疾病。 1998年3月初,是刘杨第一次发病的时间,突如其来的左眼视力模糊令刘杨不知所措,3天之内就什么都看不见了。经过治疗,刘杨的视力慢慢好转,但据刘杨说最后还是有点视神经萎缩,看东西感觉有点发白。 现在的刘杨,因为病情的不稳定无法和正常人一样生活学习和工作,尽管她已经在休养期间考取了会计师资格证等各种技能的培训证书,但是疾病的无常和身体的脆弱无法让她能够胜任任何的工作,只能在家里静养。 如今的她在经历过内心的无助和挣扎后选择了对她来说唯一的心灵寄托,那便是每周去几次基督教堂,把心中的苦闷或者是对于无法实现的梦想向牧师倾吐或者是在青年聚会的唱诗班上跟随着和她同龄的正常年轻人一起歌唱。 多发性硬化症(MS) 东亚地区患病率约为2.1~2.2/100000,是一种中枢神经系统脱髓鞘疾病,表现为下肢无力、疲劳、四肢意向性震颤、随意运动及步态的共济失调、眼球震颤、意向性震颤等,会导致神经系统受损。患者需要长期使用昂贵的干扰素等药物进行治疗。
有种勇敢叫承担 图文/梁莹菲 
龙飞在老家,看到一片绿油油的植物,他忍不住躺了上去。
从刚跟他会面开始,我就在一旁偷偷地打量这个阳光的大男孩,他的样子很秀气,尤其眼睛特别好看,但看来看去,实在没有哪里像个病人的,更别说一个罕见病人了。
这个病不像别的罕见病有让人一目了然的病征。只要得到正确诊治,患者表面上和一般人几乎一样,如果排除其他并发症的因素,其实只要一直打针吃药就能维持正常状态。但即使如此,患者大都对自己的病讳莫如深,可能他们真正的伤痛,并不在身体上,而在心灵上。 卡尔曼氏综合征(KS)是伴有嗅觉缺失或减退的低促性腺激素型性腺功能减退症。患者表现为嗅觉缺失,男性外生殖器幼稚状态,无青春期第二性征发育。女性患者内外生殖器发育不良,青春期时无乳房发育,无腋毛、阴毛,无月经。 龙飞两年前确诊此病后,每次到医院看病都要到男科转一转,向医生派发罕见病资料。他一直独自执行这些任务,看似是些无私的付出,其实也算一种自我救赎吧。 卡尔曼氏综合征(KS) 男性发病率约为1/10000,女性发病率约为1/50000,是一种具有临床及遗传异质性的疾病,表现为嗅觉缺失、无青春期第二性征发育、相关躯体发育异常。目前尚无特效疗法,患者需通过长期用药维持体征与身体稳态。
高大的身躯里住着与你我同样的灵魂 图文/孙湛 
宋杰关上阳台的纱门,由于肢端肥大症遗留下的骨质疏松导致了左腿骨折,行动不便,他的日常活动范围基本局限在家中。
“像得了一场重感冒”,痊愈后的宋杰这样告诉父母。 2009年,45岁的宋杰患上了肢端肥大症。怕父母担心,他和妻子绝口不提,直到次年1月手术成功。 即使已经治愈,宋杰还是留下了异于常人的容貌,“手足增大,皮肤增厚,颜面粗糙”。今年六月,已经痊愈的宋杰在爬楼梯时摔了一跤,肢端肥大引起的骨质疏松被放大,左腿严重骨折的宋杰不得不回崇明养伤。 患病后,宋杰加入了一个病友群,遇到了120多个“同病相怜”的病友。自费3万元治疗后,宋杰成为了病友里仅有的十余个被治愈者之一。“肢端肥大症可以治愈,但群里百分之九十的病友却因为这三万多的费用而放弃了治疗”,宋杰说。 他还说,“希望你能够把罕见病患者看作正常人一样,我们需要的不是怜悯,而是平等的对待”。 肢端肥大症(Acromegaly) 患病率约为5~7/100000,是一种内分泌及代谢性疾病,表现为体型胀大,生理功能异常。垂体肿瘤较小时可放射治疗;肿瘤较大时需手术治疗,费用昂贵。
暗夜中的光 图文/张亦蕾 
王冲去盲人按摩店上班,这是她第一次离开家,尝试独立生活。
王冲这个名字很男子气,但其实这是个高挑好看的女孩儿。她13岁查出患有白塞氏病(BD),17岁失去视力,是北京协和医院白塞氏病患者中病情最严重的一例。 在这个家庭中,女儿王冲患罕见病而眼盲,妈妈先天双腿残疾,两年前查出淋巴癌现仍未彻底脱险,爸爸因嫌弃女儿残疾而抛弃母女,妈妈独自一人靠政府补助和开残疾车抚养女儿和赡养老人,姥姥因中风而行动不便多年……在我三十多年的社会阅历中,少有家庭遭遇如此的磨难。 但王冲看起来简直是一个再健康活泼不过的90后姑娘:她喜欢戴棒球帽耍酷,会悄悄“偷”表姐的手链缠在自己手腕上臭美,会在客厅里给姥姥表演时装步逗乐,还会要求妈妈在她的新牛仔裤上故意抠几个破洞,如果感觉我的镜头在面对她,她会给我举起剪刀手。 她并不喜欢现在的这个名字,她在QQ空间里给自己起了个新名字叫李梦琪,李是妈妈的姓。
白塞氏病(BD) 又称丝绸之路病,在我国患病率约为1.4/10000,是一种慢性全身性血管炎症性疾病,表现为复发性口腔溃疡、生殖器溃疡,会有眼部、血管、神经系统病变,严重者导致失明、肠穿孔或死亡。目前尚无特效疗法。
她的快乐很奢侈 图文/胡令丰 
张艳说,第一次给悦悦打针时非常紧张,打了半个小时也没把针打进去。不过,她现在对打针已非常熟悉了。
李蒲悦,今年四岁,在出生三个月后被确诊为MMA患者。这是一种罕见的遗传代谢病,发生概率为七万分之一。它使身体无法正常代谢食物中的四种氨基酸,造成体内毒性物质增加,从而破坏神经系统和造血系统。 如果在出生后72小时内确诊并及时治疗,有可能避免脑损伤等情况。只需定期打针吃药,便能和正常的小孩一样健康长大。但由于MMA没有被纳入我国的新生儿筛查范围,许多小孩在发病后才被确诊。悦悦在发病后三个月才确诊,已经患有了严重的酸中毒和脑损伤。现在,四岁半的悦悦智力只相当于三个月大的婴儿,患有重度残疾,无法说话,无法站立,也看不见东西。 MMA患者每周都需要注射B12针剂。因利润低,这种针剂没有一家国内药厂生产。悦悦的父亲常委托国外的客户从德国带药,一带好几十盒,以原价70元的价格分给病友。他多次联系了广东省卫生厅和药监局,表达自己的诉求,一是将MMA纳入新生儿筛查,防止更多悲剧的发生;二是国内药厂为罕见病患者生产孤儿药,无奈一直得不到正面回复。
甲基丙二酸血症(MMA)
发病率约为1/50000,是一种常染色体隐性遗传代谢性疾病,表现为生长发育不良,智力落后或倒退,新生儿期发病呕吐、脱水、厌食、呼吸急促甚至昏迷、死亡,急性期治疗以补液、纠正酸中毒为主,必要时需透析。目前尚无特效疗法,患者需要长期的营养治疗和药物治疗。
勇气是他的战袍 图文/吴育琛 
陈勇双手的特写,显得苍白,没有血色。
“阵发性睡眠性血红蛋白尿。”——陈勇一口气念出他所患疾病的全称,11个字,不带一点停顿。 08年,在从湘雅医院的一个医生口中第一次听到这个冗长病名之前,他一直以为自己患的是先天性心脏病、贫血得比较厉害。这个病的名字不太好记,5年来,陈勇一次次地对不同人念出这个名字,有时是为了描述病情,有时是为了借钱看病,有时是为了输血救命,有时则是为了寻找同伴。久而久之,这个词成了他的潜意识的一部分,能够不假思索地脱口而出,就像他自己的名字一样。 因为身体的原因,他的脸色看起来很差,不能干重活,很容易疲劳,他没有文凭,因为生病休学以后,学校认为他的身体状况不足以完成学业,没有公司愿意聘用他。他只得终日徘徊在家附近大桥底下的江边,无所事事,人生仿佛停滞,无法前行。 阵发性睡眠性血红蛋白尿症(PNH) 发病率约为2.8/1000000,是一种获得性克隆性疾病,表现为重度贫血、血栓、肺动脉高压、器官衰竭。国外已有针对该疾病的药物但价格十分昂贵。目前国内暂无针对性治疗药物,除了输血,几乎没有治疗手段。
蝴蝶有爱不孤单 图文/钟锐钧 
妞妞下楼玩的时候骑车摔倒了,妈妈赶紧上前安慰女儿。
妞妞的样子和普通的孩子并没有很大区别。她很漂亮,大眼睛,胖乎乎的很结实,性格开朗活泼,偶尔也会有些小脾气。 然而,只要和她相处几天,便会发现她有那么一点的不同: 说话偶尔会咬字不清晰,大动作的运动能力显得稍弱,仔细看妞妞的皮肤,可以发现她身上有一点点斑痕。 妞妞的全名庞雅雯,她今年4岁。她患有结节性硬化症,简称TSC,一种常染色体显性遗传的神经皮肤综合征。 从妞妞确诊的时候起,妈妈袁碧霞查找了大量的资料,找到了跟孩子同病相连的病友群。袁碧霞发现,TSC的病友情况都很不一样,有的长大成人后读书,参加工作,严重的生活不能自理,失明等等。李惠颜是前者,她已经参加工作,在药物的帮助下,过着和正常人一般的生活。如果不是脸上的蝴蝶斑,一般人很难会察觉她患有疾病。 对袁碧霞来说,情况好的病友,就是妞妞的希望之光。 结节性硬化症(TSC) 发病率约为1/10000,是一种常染色体显性遗传病,表现为面部皮脂腺瘤、癫痫发作和智能减退,可能导致肾功能衰竭、心力衰竭、癫痫持续状态、呼吸衰竭等并发症。目前尚无特效疗法。
生命不能承受之重 图文/张星海 
晓迪的父亲是得脊髓小脑共济失调去世的,他的病也是从父亲这里遗传来的。
“每个人都有自己的命运,我的命运就是这个病。既然被安排成这样,所以无所谓了。”说这话的人叫余晓迪,今年29岁。他说话时口齿不清,不能行走,整天只能坐在轮椅上。 他并不是一般的残疾人,他患有一种非常罕见的疾病脊髓小脑性共济失调。患者大脑基本不受影响,意识清晰、思维正常,然而躯体逐渐被禁锢,最终因器官功能衰退、丧失而失去生命。因为刚发病的时候患者步态不稳、走路像企鹅一样摇摆不定,被叫做“企鹅人”。 晓迪有哮喘,这和他患的共济失调这个病有很大关系。如果哮喘没有发作,他和普通坐轮椅的残疾人没有什么差别。但哮喘发病时却很是吓人,他的嗓子里会发出长长的声音,气一口接上不一口,嘴里会吐出一堆痰或者刚刚吃进去的食物。而且这时的他对气味也非常敏感,大街上汽车的尾气或者饭馆里飘出的油烟气味都会使他非常难受。遇到这种时候,他只能逃离或者赶紧去医院吸氧,但到了医院,病房的气味也依然使他难受。 脊髓小脑性共济失调(SCA) 全球患病率约为3/100000,是一种常染色体隐性遗传病,表现为共济失调、弓形足、视神经萎缩、脊柱侧弯、心肌病、癫痫发作、视力减退或丧失,心脏损害等。目前尚无特效疗法。该群体也被称为“企鹅人”。
你永远不能低估一颗想奔跑的心 图文/崔楠 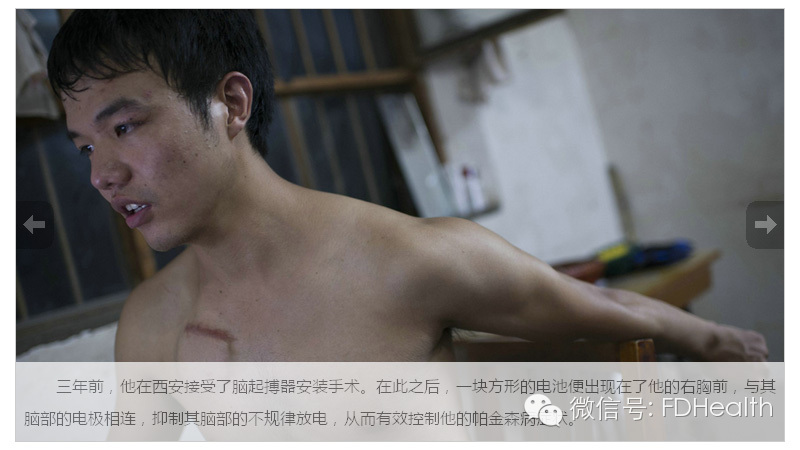
三年前,他在西安接受了脑起搏器安装手术。在此之后,一块方形的电池便出现在了他的右胸前,抑制其脑部的不规律放电,从而控制他的帕金森病症状 。
焦飞与哥哥相差六岁,不幸的是兄弟两个均罹患青少年型帕金森病。虽然帕金森这种病本身借由美国拳王阿里的缘故并不让人感到陌生,但发病于孩童时期的青少年型帕金森症患者却罕有发现。而不满十岁时便开始发病的焦飞是有记录以来中国发现的最早的,也是世界范围内,在极幼小年龄中便开始发病的第四例病患。 9月的中秋,我第一次见到了他。惊讶于他强壮的身体,若不是在他扯开衣领后,看到其右侧胸口那一道深红的刀疤,以及胸肌上那块明显突起的脑起搏器电池,也许没有任何人相信站在自己面前的这个健壮少年已是一名饱经病痛侵袭10余年的青少年型帕金森病人。 由于哥哥在焦飞小时候已经被确诊为青少年型帕金森,焦飞身上逐渐的变化也让还是孩子的他慢慢意识到或许自己也会变成哥哥“那个样子”,而这对一个从小就立志成为一名侦察兵的他来说,无疑相当于一记击碎梦想的铁拳。 青少年型帕金森病(YOPD) 占帕金森发病总人数的10%,是一种神经系统变性疾病,表现为静止性震颤、肌强直、运动迟缓、姿势步态障碍,易伴有智力障碍和发生动眼危象。目前尚无特效疗法。
十米长的呼与吸 图文/商华鸽 
开着代步车到人少的地方后,王晓莉会戴上氧气管吸两口。她怕小区里的街坊邻居看到自己戴氧气管的样子。
王晓莉成名已久。 那是十年前的事了。2003年,北京协和医院,河南郑州人王晓莉被确诊为肺淋巴管肌瘤病(LAM)。她是协和医院被确诊的第三例患者,当时全国也不过五例。十年后王晓莉回忆起当时自己住院的时光,仍记得自己走在楼道里似乎也趾高气扬,“我在医院里可有名了,很多医生经常都来看我。毕竟是很罕见的病例,是多好的研究对象。” 十年来,国人对罕见病“LAM”的了解仍然粗浅:它只在育龄期女性身上发病,发病起因不明,呼吸会极度困难,目前无法根治,晚期要做肺移植,患病几率在百万分之一。问题的另一面在于,目前国内也仅有北上广屈指可数的几家医院设有专门的LAM门诊。甚至不少医生,仍并未听说过这种病——被误诊为气胸,自然再正常不过。 王晓莉第一次见面时就对我说,自己真的是中彩票了。她十年前了解到,LAM患者从发病到去世一般不超过十年。十四年了,这朵喘息的女人花仍在坚忍开放。 肺淋巴管肌瘤病(LAM) 发病率约为1/400000,是一种弥漫性肺部疾病,表现为气胸、乳糜胸、慢性进展的呼吸困难,严重者会导致死亡。目前尚无特效疗法。
离地面更近,自有别样的风景 图文/张星海 
帝刚的作息和大多数人不同。每天傍晚,是都市人下班回家的时候,但对他来说,却是去上班的时间。
站在记者面前的濮阳希刚像个袖珍人,身高一米三左右,面带笑容,声音稚嫩。其实他是一种罕见病患者,这种疾病的医学名称叫生长激素缺乏症,主要的特征就是个头矮小,面容幼稚,始终像一个长不大的孩子。
在某档综艺节目中,完整的记录了一段濮阳希刚向一个比他矮的袖珍女孩真情表白的片段。最后两人紧紧的拥抱在一起。女孩说:“希望我们能幸福吧!”希刚回答:“我们必须幸福!” 这段视频非常感人,但希刚说,里面的许多桥段都是编排出来的,事实上他们俩早已经确立恋爱关系。但让人遗憾的是,这段感情最终没有开花结果,女孩后来回了东北,他们就此彻底分开了。希刚后来到一家KTV上班,工作是向客人推销冰激凌和酸奶,有时也会唱歌,客人会给些小费。这样的工作虽然不是希刚喜欢的,但相对还算稳定。 袖珍人的父母兄弟差不多都是正常人,所以在他们生长的坏境中,他们是唯一奇怪的人。在没有来大城市之前,他们甚至都只认为世界上只有自己是这样的矮小的人。 生长激素缺乏症(GHD) 发病率约为1/8500,是一种内分泌疾病,表现为身材矮小、生长障碍、性发育障碍、代谢紊乱、神经系统、心、肾功能异常。目前一般使用基因重组人生长激素治疗。该群体也被称为“袖珍人”。
以上图文均节选自中国罕见病发展中心网站。
| 




 鲁公网安备 37020202001532号
鲁公网安备 37020202001532号 







 IP卡
IP卡 狗仔卡
狗仔卡

 提升卡
提升卡 置顶卡
置顶卡 沉默卡
沉默卡 喧嚣卡
喧嚣卡 变色卡
变色卡 抢沙发
抢沙发 千斤顶
千斤顶 显身卡
显身卡